

شرکت علوم زیستی شرودینگر، از قدیمیترین شرکتهای خدماتی کامپیوتر در حوزه علوم زیستی است. شرکتی که در سال 1990 توسط دو نفر شیمیدان تاسیس شد و در سالهای ابتدایی به عنوان تسهیلگر تحقیق در زمینه تحقیقات علومزیستی فعالیت میکرد. با توسعه نقش محاسبات در علوم زیستی بخصوص شیمی و مولکولی از اوایل سال 2000 این شرکت همکاری بسیار نزدیکی با تیمهای توسعهدهنده نرمافزارهای محاسباتی ایجاد کرد. حدود سال 2010 و با تاسیس شرکت علوم درمانی Nimbus Therapeutics فعالیتهای این شرکت نقش مهمی در توسعه و درک نحوهی ایجاد و درمان بیماریها شروع کرد.
یکی از اولین نتایج تحقیقات شرکت نیمباس کشف و ساخت بازدارنده ACA (استیل کوآ کربوکسیلاز) است که در سال 2016 به قیمت 1.2 میلیارد دلار به شرکت داریویی 90 میلیارد دلاری گیلیاد فروخته شد. این بازدارنده به زودی به عنوان داروی موثر اختلالات کبدی وارد بازار خواهد شد. تاسیس شرکت نیمباس در سال 2010 منجر به اولین دور سرمایهگذاری شخص بیلگیتس در شرکت شرودینگر شد.
با شروع کرونا و مشخص شدن نقش سرعت و زمان در تحقیقات مولوکولی، محبوبیت خدمات شرکت شرودینگر و بستهی نرمافزاری قدیمی و معروف این شرکت Schrodinger Suite به شدت افزایش پیدا کرد. در اواخر سال 2019 آخرین دور سرمایهگذاری خصوصی این شرکت به مبلغ 108 میلیون دلار انجام شد و زمزمههای ورود شرکت 30 ساله شرودینگر به بازار سهام آغاز شد.
ورود شرودینگر به بازار سهام – NASDAQ: SDGR
در فوریه 2020 این شرکت در نهایت وارد بازار سهام شد و در پایان اولین روز عرضهی اولیه سهام، قیمت هر واحد سهم این شرکت به 27.22$ رسید. با ورود به بازار سهام، شرودینگر توانست 232 میلیون دلار سرمایه جذب کند. ارزش بازار این شرکت در حال حاضر (شهریور 1400) حدود 4.15 میلیارد دلار است و قیمت سهام آن در بازار سهام نزدک حدود 55$ در ازای هر سهم است. درآمد این شرکت در سال 2018 تنها 66.6 میلیون دلار بوده است، اما با گسترش تحقیقات برای داروهای مولکولی و ورود مشتریانی مانند سانوفی و فایزر روند روبهرشد به سرعت ادامه دارد. پیشبینی میشود اندازه بازار تحقیق و توسعه در صنایع دارویی در سال 2022 حدود 202 میلیارد دلار باشد. معمولا نقش شرکت Schrodinger در صنایع علوم زیستی را با شرکت تسلا در صنایع خودروسازی مقایسه میکنند.
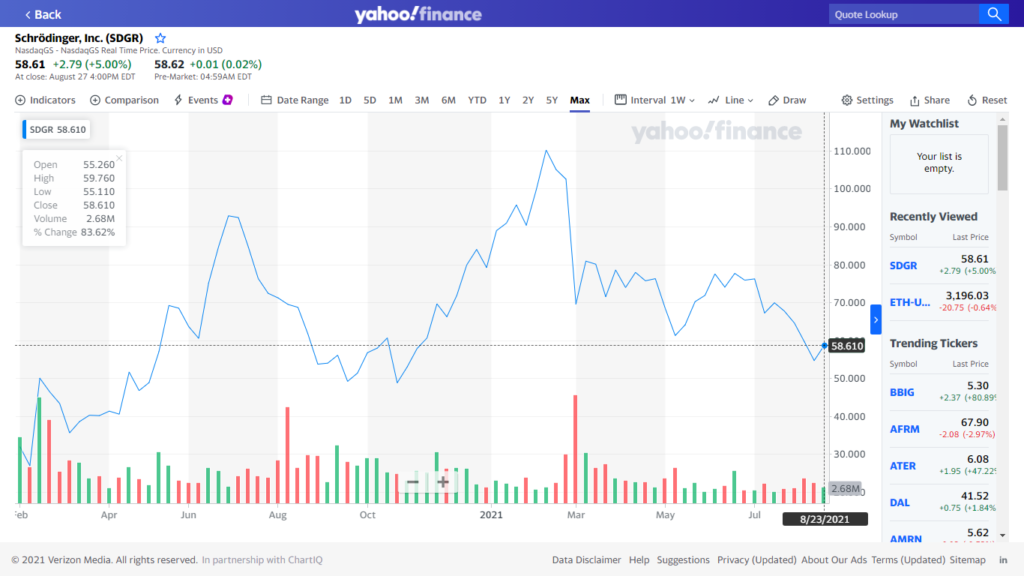
مهمترین ابزار ایجاد تحول در علوم زیستی و مولکولی شرودینگر بستهی نرمافزاری Schrodinger Suite است که در تعامل نزدیک با شرکتهای تولیدکننده نرمافزار و شرکتهای دارویی و تحقیقاتی توسعه داده شده است. این شرکت، با انعقاد قراردادهای حق استفاده مختلف و اختصاصی بین طرفین، امکان استفاده در لایههای مختلف را بین مراکز تجاری، تحقیقاتی و دانشگاهی فراهم میکند. همچنین با استفاده از پلتفرم ایجاد شده، امکان سرعت بخشیدن به محاسبات و حل مشکلات را فراهم میکند.
شرودینگر از سال 2012 توانسته با همکاری آمازون سرعت حل و کشف مسائل مربوط به بیماریها و درمان را سرعت بخشد. به عنوان مثال در سال 2012 با استفاده از سیستم Cycle Computing توانست در مدت سه ساعت و با استفاده از 50هزار هسته پردازشی یکی از مشکلات را حل کند. Cycle Computing هم اکنون در اختیار مایکروسافت است.
در بهار 1399 شرودینگر و گوگل وارد یک برنامه سه ساله مشارکتی شدند تا با استفاده از قدرت Google Cloud برای کشف سریعتر داروها اقدام کنند. در ادامه تصویری از بستهی نرمافزاری Schrodinger Suite نسخه 2021 را میبینید. همچنین بستههای مختلف این پکیج برای استفاده تحقیقاتی دانشگاه استنفورد فهرست شده است.
Biologics Suite
Comprehensive Protein Modeling
- Complete set of homology modeling tools, including both rapid and advanced methods
- Chimeric and multimeric models
- Advanced sequence tools for multiple alignment, with extensive annotation options
- Protein structure quality analysis
- Identification of consensus elements in structural families/homologs
Advanced Features for Protein Engineering
- Protein aggregation prediction
- Identification of hot spots for proteolysis, glycosylation, deamidation, and oxidation
- Residue-based analysis of energies, solvent-accessible surface areas, and hydropathy
- Cysteine scanning to identify residue mutations for potential cysteine-cysteine disulfide bonds
- Automated residue scanning to predict relative stabilities as well as changes in solvent‑accessible surface area, pKa, and hydropathy
Antibody Modeling
- Automated intuitive workflow
- Prediction of CDR from sequence
- Rapid prediction using curated antibody database
- Advanced ab initio loop prediction using Prime
- Database management tools for simple incorporation of new/proprietary structures, and allows use of multiple databases in modeling
State-of-the-Art Protein-Protein Docking
- Well-validated docking code, PIPER; for more information please visit the PIPER website.
- Special antibody and multimer modes
Advanced Simulations
- Access to Schrödinger simulation tools
- Advanced molecular dynamics (MD)
- Extensive Free Energy Perturbation (FEP) tools
- Large-scale, low mode (normal) search for domain movement
- Quantum mechanics/molecular mechanics (QM/MM) predictions of binding site reactivity
Small-Molecule Drug Discovery Suite
Wide Range of Virtual Screening Options, Spanning the Spectrum of Speed vs. Accuracy Tradeoffs
- 2D/3D QSAR with a large selection of fingerprint options
- Shape-based screening, with or without atom properties
- Ligand-based pharmacophore modeling
- e-Pharmacophore modeling incorporating ligand-receptor interaction energies
- Flexible ligand docking with industry-leading Glide
- SIFt — structure interaction fingerprint analysis
- Induced-fit docking with receptor flexibility
- Covalent docking
- 2D ligand interaction diagrams
Advanced Computations to Estimate Binding Affinity and to Rank-Order Compound
- Embrace post-docking refinement
- Prime MM/GBSA
- Free energy perturbation (FEP) theory
- Linear interaction approximation (LIA)
- QM-polarized ligand docking
Analyses to Predict, Prepare, Refine and Characterize Target Structureand Binding Modes
- Protein crystal structure refinement
- Protein structure analysis and homology modeling
- GPCR and hERG modeling
- Protein binding site identification and analysis
- Multiple binding mode prediction
Complete Set of Utilities to Prepare, Analyze and Filter Ligand Structures and to Create and Design Ligand Libraries
- 2D to 3D structure conversion, with emphasis on bioactive conformers
- Tautomeric state enumeration and analysis
- Ligand interaction diagram
- Commercially-available compound database
- Flexible ligand superposition
- Combinatorial library creation
- Core hopping
- Filter compound libraries based on predicted ADME properties
- R-group analysis
General Modeling Tools That Can be Applied Across a Wide Range of Chemical Systems
- High-performance QM calculations, in gas phase and in solution
- MM/MD simulations, with implicit or explicit solvents
- Small molecule and macromolecular conformational analyses
- Mixed-mode QM/MM calculations for ground state and reactivity studies
Fully Supported by State-of-the-Art Visualization and Workflow Automation Tools
- Unified graphical user interface, Maestro, that serves all computations
- Publication-quality graphics and flexible analysis
- KNIME Extensions and customizable workflows
- Python API
دیدگاهتان را بنویسید